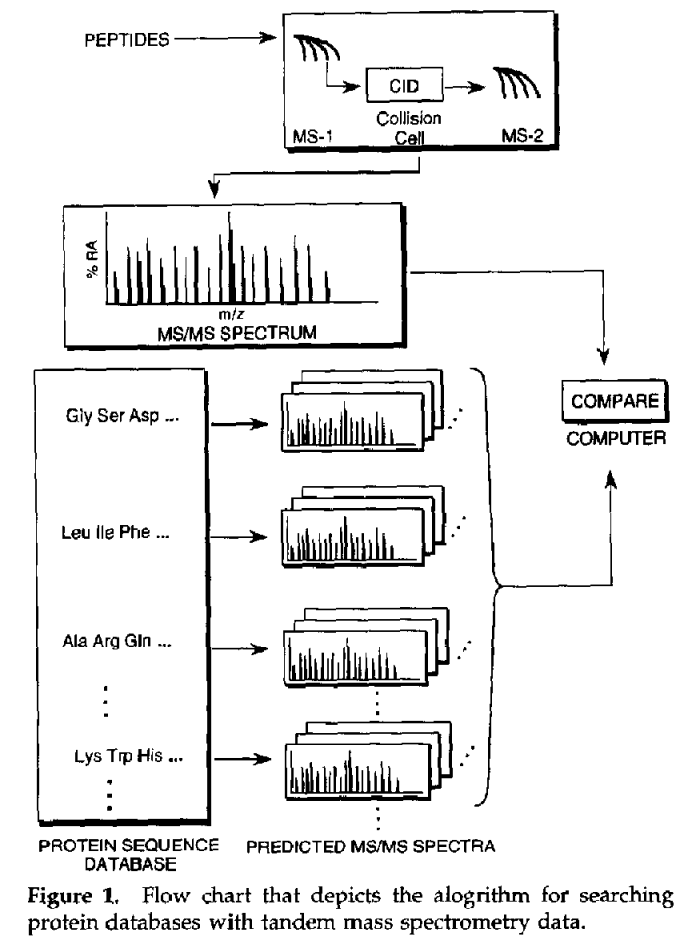
At ProGenTomics, protein identification can be performed on a TripleTOF5600 (Sciex), a SynaptG2Si (Waters), a TripleTOF6600 (Sciex) and a ZenoTOF7600 (Sciex) by both data-dependent acquisition (DDA) and data-independent acquisition (DIA). DDA has been the most widespread strategy for protein identification for over two decades. However, currently the more data-rich and comprehensive DIA is taking over the field.
During data-dependent acquisition, the instrument selects an amount of targets for fragmentation per cycle based on predefined characteristics such as charge state, intensity, isotope distribution, prior selection… This implies that the selection of peptides is intrinsically stochastic and can cause missing values.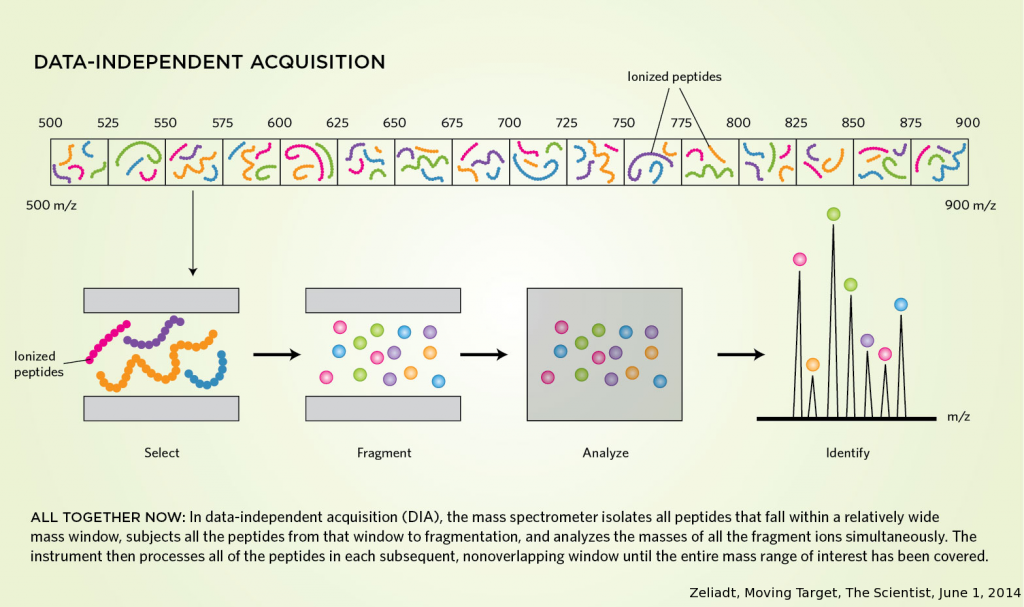
Data-independent acquisition on the other hand is a term that covers all acquisition strategies without prior knowledge or data based precursor selection. This not only avoids the stochastic selection of peptides for fragmentation – which is rarely enough to cover all analytes of a complex sample – but also allows for quantification at both the fragment and precursor level. This in turn gives DIA a better discriminating power when dealing with ambiguity or post translational modifications of peptides with the same backbone. However, the biggest drawback are the shear complexity that comes with the simultaneous fragmentation of multiple peptides and the shortcomings of certain softwares that can not identify several PTMs or non-tryptic peptides.